Distrofia miotónica
Qué es la distrofia miotónica
La distrofia miotónica (DM) es una forma de distrofia muscular que afecta a músculos y muchos otros órganos del cuerpo. La DM es una enfermedad muscular rara que solo afecta a 1 de cada 8.000 individuos y se divide en dos tipos principales según la alteración genética que causa la enfermedad.
¿Qué es una enfermedad rara? Una enfermedad rara es aquella que afecta a un pequeño número absoluto de personas o a una proporción reducida de la población. En Europa, se considera «rara» a una enfermedad que afecta a menos de 1 de cada 2.000 personas. En Estados Unidos, se define así a un trastorno o enfermedad que sufren menos de 200 000 personas.
Distrofia miotónica de Steinert o distrofia miotónica tipo 1
La distrofia miotónica tipo 1 (DM1 o enfermedad de Steinert, OMIM: 160900) es la más común. La DM1 es un trastorno de patrón autosómico dominante causada por una expansión de trinucleótidos inestable de la citosina-timina-guanina [CTG] n ubicada en la región 39 no traducida del cromosoma 19q13.3. En la literatura se publican excelentes revisiones sobre el origen genético de la distrofia miotónica tipo 1 DM1 y el papel del ARN tóxico en la patogénesis.
La expansión [CTG] n responsable de DM1 puede variar de 50 a más de 1000 repeticiones y contribuye a una amplia gama de fenotipos diferentes. De acuerdo con el International Myotonic Dystrophy Consortium (IDMC) (Neurology, 2000), se reconocen tres fenotipos clínicos diferentes en la distrofia de Steinert DM1 según la edad de inicio junto con las repeticiones de [CTG] n:
-Suave: 50 - 150 repeticiones CTG
-Formas clásicas (adultas) y de inicio tardío: 100 - 1000 repeticiones de CTG
-Congénita: > 1000 repeticiones CTG
Qué síntomas tiene la enfermedad de Steinert
Clínicamente, la distrofia miotónica tipo 1 (DM1) se caracteriza por una atrofia y debilidad muscular progresiva, miocardiopatía, resistencia a la insulina y cataratas, entre otros. La distrofia miotónica DM1 o enfermedad de Steinert afecta a mujeres y hombres por igual.
Avances destacados de la enfermedad de Steinert en la I+D de Myogem
Actualmente, no hay cura para los pacientes con la enfermedad de Steinert DM1, sino tratamientos paliativos de la sintomatología, por lo que existen múltiples necesidades médicas no cubiertas en este colectivo de enfermos. El manejo es principalmente sintomático, enfocado en la rehabilitación, el apoyo psicológico y la monitorización regular mediante ECG para detectar alteraciones en la conducción cardíaca. Es crucial realizar un estudio de todos los familiares en riesgo y proporcionar un adecuado consejo genético, incluyendo la recomendación de diagnóstico prenatal en embarazos de madres afectadas.
Según los datos obtenidos por Myogem Health Company a partir de los experimentos in vitro e in vivo realizados durante los últimos diez años, el producto MYODM ha demostrado su capacidad para aumentar los niveles de expresión de MBNL, tanto MBNL1 como MBNL2 y su cantidad efectiva en el núcleo de mioblastos humanos DM1.
Los principales ingredientes de MYODM son las metilxantinas teobromina y cafeína y ningún otro alimento contiene la proporción de la formulación en MYODM. Las metilxantinas son metabolitos secundarios de plantas derivados de nucleósidos purínicos y están presentes en casi 100 especies diferentes de plantas (entre ellas, Theobroma cacao, Coffea arabica y Camelia sinensis).
MYODM contiene 20mg de cafeína por cápsula, por lo que la toma diaria de tres cápsulas equiespaciadas conlleva una ingesta máxima de 60mg. En este aspecto, MYODM cumple con la legislación vigente en materia de contenido de cafeína en complementos alimenticios, para los que se aceptan como máximo una cantidad diaria de 80mg.
Tomando como referencia a un individuo de 70kg de peso, la toma de una cápsula de MYODM (con sus 20mg de cafeína) equivaldría a una ingesta de 0.28mg/kg en un individuo de 70 Kg de peso. La Agencia de Seguridad Alimentaria Europea (EFSA), en sus informes de expertos ("Scientific Opinion on the safety of caffeine", EFSA Journal 2015;13(5):4102) indica que, en la población sana en general, son seguras dosis individuales de cafeína de hasta 3 mg/kg para un adulto de 70 Kg (equivalente a 200 mg de cafeína en una sola ingesta). En general, dosis habituales de cafeína hasta 400 mg/día se consideran seguras en adultos.
¿La distrofia miotónica de Steinert es hereditaria?
La distrofia miotónica es una enfermedad hereditaria. Se transmite de padres a hijos a través de un patrón autosómico dominante, lo que significa que si uno de los padres tiene la enfermedad, hay un 50% de probabilidad de que sus hijos la hereden, tal y como se recoge en 'Estudio epidemiológico de la distrofia miotónica congénita de Steinert: Características dismorfológicas'.
En el caso específico de la distrofia miotónica de Steinert, se hereda de manera autosómica dominante a través de la madre. Esto significa que si la madre tiene la enfermedad, existe una alta probabilidad de que sus hijos también la tengan.
Diagnóstico de la distrofia miotónica o síndrome de Steinert
Pese a que la enfermedad de Steinert se manifiesta principalmente en los músculos esqueléticos, los primeros síntomas pueden no ser musculares, lo que supone un retraso en el diagnóstico. Por ello, es fundamental realizar un estudio genético en todos los familiares de riesgo para identificar a los portadores y proporcionar un consejo genético adecuado. Más allá de la debilidad muscular, la atrofia o la miotonía, estos son algunos otros aspectos clínicos por los que se puede manifestar la enfermedad:
·Cardíacos: arritmias debidas a fibrosis de las vías de conducción, que pueden incluir bloqueo auriculoventricular de primer grado, bradicardia sinusal y bloqueo de rama.
·Respiratorios: hipoventilación por hipotonía diafragmática y riesgo aumentado de infecciones respiratorias.
·Otros: cataratas (frecuentes y usadas como marcador de portador), calvicie frontal en varones, atrofia gonadal, alteraciones gastrointestinales gastrointestinales como disfagia y dolor abdominal inespecífico, y problemas metabólicos como la alteración de la glucemia.
¿Qué estudios complementarios se recomiendan para la detección o diagnóstico de la distrofia miotónica?
·Electrocardiograma (ECG): para detectar arritmias y otras alteraciones en la conducción cardíaca.
·Análisis de sangre: puede revelar elevación de las CPK musculares.
·Análisis genético del ADN: permite el diagnóstico prenatal y la identificación de familiares asintomáticos o con pocos síntomas.
·Estudio neurofisiológico: muestra un patrón miopático y es útil para el diagnóstico precoz y el seguimiento de la enfermedad.
¿Afecta la enfermedad de Steinert a la esperanza de vida?
Los síntomas de la distrofia miotónica tipo 1 avanzan de forma progresiva, lo que puede mermar de forma significativa la esperanza de vida de los pacientes. Algunas de las complicaciones asociadas a la enfermedad son problemas del sueño como hipersomnia diurna o somnolencia excesiva durante el día, dificultades cardiovasculares como la miocardiopatía o arritmias u otros síntomas como la caída de los párpados, cataratas, calvicie precoz en varones o diabetes.
Es por todo esto que la esperanza de vida de los pacientes con enfermedad de Steinert puede verse significativamente reducida debido a las complicaciones mencionadas. Generalmente, no supera los 55 años en muchos casos debido a la progresión de la debilidad muscular y los problemas cardíacos.
Sin embargo, la extensión del impacto en la esperanza de vida depende en gran medida del momento en que aparecen los síntomas. La monitorización constante y el manejo de los síntomas pueden mejorar la calidad de vida y potencialmente extender la esperanza de vida dentro de los límites que la enfermedad permite.
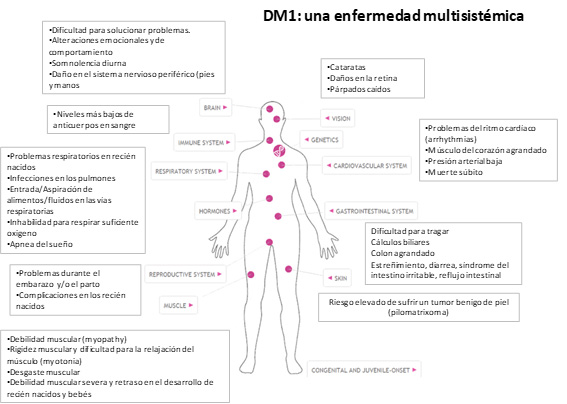
Figura adaptada de Myotonic Dystrophy Foundation
